介绍
腓骨肌萎缩症(Charcot-Marie-Tooth,CMT)亦称为遗传性运动感觉神经病(HMSN),具有明显的遗传异质性,临床主要特征是四肢远端进行性的肌无力和萎缩伴感觉障碍。CMT是最常见的遗传性周围神经病之一(发病率约为1/2500)。根据临床和电生理特征,CMT分为两型:CMT1型(脱髓鞘型),神经传导速度(NCV)减慢(正中神经传导速度s),CMT2型(轴突型),神经传导速度正常或轻度减慢(正中神经传导速度>38m/s)。多数呈常染色体显性遗传,也可呈常染色体隐性或X-连锁遗传。
最新文章
症状
I型(脱髓鞘型)
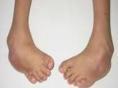
是典型的腓骨肌萎缩症双下肢呈倒立酒瓶状或称鹤立腿同时出现足弓高耸爪形趾马蹄内翻畸形等行走时表现特殊的跨越步态表现肌无力肌萎缩肌束颤腱反射减退或消失首发于手部肌前臂肌萎缩而后下肢远端肌萎缩者仅见于少数病例四肢末梢可出现手套状袜状深浅感觉障碍和一系列植物神经与营养代谢障碍局部皮肤呈青紫色皮肤温度低溃疡形成等。
II型(轴突型)
发病晚成年开始有肌萎缩部位和症状与I型相似但程度较I型为轻临床上按症状组合有多种变异型如肩胛腓骨肌萎缩型视神经萎缩型弗里德赖希共济失调若伴有腓骨肌萎缩则称腓骨肌萎缩型共济失调即Roussy-Levy综合征。
病因
遗传因素
CMT1A致病基因定位于17p11.2-12,核基因编码周围神经髓鞘蛋白22(PMP22),PMP22基因重复突变导致过度表达,使PMP22蛋白增加;小部分病人因PMP22基因突变,产生异常PMP22蛋白而致病。
检查
本病的辅助检查方法主要是神经电生理检查
神经传导速度的检测对该病的诊断有很大的帮助对分型则必不可少常选大小鱼际肌胫前肌腓肠肌进行肌肉安静状态下有无矢神经电位小力收缩时运动单位电位的时限波幅大力收缩时的电位位相及峰值电位检测用表面电极检测运动神经传导速度(motornervecon2ductionvelocity,mcv)及感觉神经传导速度(sensorynervecon2ductionvelocity,ncv)常检测正中神经和尺神经的mcv及scv胫神经和腓总神经的mcv腓肠神经的scv肌电图神经传导速度正常值参照汤晓芙标准大多数患者可表现为均匀一致的减慢而无传导阻滞构成遗传性脱髓鞘神经病的特征性表现。
鉴别
1、远端型肌营养不良:四肢远端逐渐向上发展的肌无力,肌萎缩,该病成年起病,肌源性损害肌电图,运动NCV正常等可以鉴别。
2、慢性进行性远端型脊肌萎缩症(chronicprogressivedistalspinalmuscularatrophy):该病的肌萎缩和肌无力以及病程经过类似CMT病,但感觉功能不受累,EMG显示为前角损害。
3、遗传性共济失调伴肌萎缩:又称Roussy-Lévy综合征,儿童期起病,缓慢进展,表现腓骨肌萎缩,弓形足,脊柱侧凸,四肢腱反射减弱或消失,运动NCV减慢,但有站立不稳,步态蹒跚,手震颤等共济失调表现。
并发症
本病常有肿胀,紫绀,溃疡等神经营养障碍,偶见视神经萎缩,瞳孔改变,眼球震颤及三叉神经痛,后期手部出现骨间肌,大、小鱼际肌萎缩,形成猿手畸形,但萎缩一般不超过肘关节以上,腿部的损伤先从腓肠肌开始,后逐渐扩展至骨间肌,小腿屈肌,最后累及大腿下1/3肌肉,但其上部完全正常,形成“鹤腿”或倒置的酒瓶样畸形,萎缩肌肉可有肌束震颤,跟腱反射早期减弱或消失,因足背屈无力常呈马蹄内翻畸形。
治疗
本病目前尚未无特殊有效治疗,主要是对症和支持疗法,垂足和足畸形可穿着矫鞋,由于病程进展缓慢,大多数患者可存活数十年,对证治疗可提高患者生活质量。
预防
腓骨肌萎缩症是遗传性运动感觉神经病,具有明显的遗传异质性,是一组遗传性疾病。腓骨肌萎缩症唯一有效的预防方法是进行产前的基因诊断。通过基因诊断确定先证者基因型,用胎儿绒毛、羊水或脐带血分析胎儿基因型,确定产前诊断并终止妊娠。
