介绍
大疱性表皮松解症(epidermolysisbullosa,EB)分为遗传性(先天性)和获得性(epidermolysisbullosaacquisita,EBA)两种。遗传性EB依据发病部位不同可分为三类:①单纯性大疱性表皮松解症(simplexEB,EBS),水疱在表皮内;②交界性大疱性表皮松解症(junctionalEB,JEB),水疱发生于透明层;③营养不良性大疱性表皮松解症(dystrophicEB,DEB),水疱发生在致密下层。
最新文章
症状
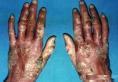
各型大疱性表皮松解症的共同特点是皮肤在受到轻微摩擦或碰撞后出现水疱及血疱,好发于肢端及四肢关节伸侧,严重者可累及机体任何部位。皮损愈合后可形成瘢痕或粟丘疹,肢端反复发作的皮损可使指趾甲脱落。
单纯型仅累及肢端及四肢关节伸侧,不累及黏膜,皮损最表浅,愈后一般不留瘢痕。营养不良型可累及任何部位(包括黏膜),病情多较重,常在出生后即出现皮损,且位置较深,愈合后遗留明显的瘢痕,肢端反复发生的水疱及瘢痕可使指趾间的皮肤粘连,指骨萎缩形成爪形手;口咽部黏膜反复溃破、结痂可致张口、吞咽困难,愈后不佳。交界型罕见,出生后即出现广泛水疱、大疱及糜烂面,预后差,多在2岁内死亡。
EBA多发生在老年人,皮损好发生在手指、足、肘膝关节侧面,容易受外伤的部位,皮损为无炎症反应的皮肤上形成水疱、大疱、糜烂等损害,愈后可留萎缩性瘢痕,可见粟丘疹,部分病人伴有毛发、甲损害,以及黏膜损害。
病因
EBS与编码角蛋白5和14的基因突变有关;JEB与编码板层素5、ⅩⅦ型胶原(BPAG2)等物质的基因突变有关;DEB与编码Ⅶ型胶原的基因(COL7A1)突变有关。由于编码表皮和基底膜带结构蛋白成分的基因突变,使这些蛋白合成障碍或结构异常,导致不同解剖部位水疱的产生。
EBA患者是一种自身免疫性疾病。病因不明,有研究发现与体内产生抗Ⅶ型胶原抗体和HLA-DR2阳性有关。
检查
单纯性大疱性表皮松解症的分子病理生理学:角蛋白多肽的突变位点与单纯性大疱性表皮松解症的严重性之间有密切关系,D-M型角蛋白突变位于多肽中央螺旋杆区的氨基(1A)或羟基(2B)端,K型突变的位置较倾向于杆区的中央部分,W-C型突变位置经常或者位于杆区的非螺旋连接(L12)区,或位于K5的前端。
鉴别
EB应与大疱性类天疱疮、天疱疮进行鉴别。
并发症
1.营养不良型大疱性表皮松解症的隐性遗传型,严重型(ITS-RDEB)最严重的合并症是在慢性糜烂区域发展为鳞状细胞癌,高于50%的TTS-RDEB患者在30岁左右时发展为此癌,许多死于癌转移。
2.交界型大疱性表皮松解症(JEB)的Herlitz型,常合并有气管水疱,狭窄或阻塞,声音嘶哑是早期婴儿恶化的征兆,显着的生长迟缓和顽固性混合性贫血使治疗更加困难,患儿常死于败血症,多器官衰竭和营养不良。
治疗
目前尚无特效疗法。应保护皮肤,防止摩擦和压迫,可用非粘连性合成敷料、无菌纱布或广谱抗生素软膏防治感染,同时应加强支持疗法。
预防
保持良好的心态非常重要,保持心情舒畅,有乐观、豁达的精神、坚强战胜疾病的信心。
