介绍
隐性脊柱裂是隐性椎管闭合不全中最为多见的一种,多见于腰骶部,有一个或数个椎骨的椎板未全闭合,而椎管内容物并无膨出。绝大多数的隐性脊柱裂终身不产生症状,也没有任何外部表现。
最新文章
症状
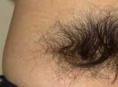
隐性脊柱裂的症状主要因受累节段的脊髓与脊神经损害引起,即与是否受压和神经损害的程度相关。局部皮肤可有毛发增多,皮肤向内凹陷,有的呈现不规则的毛细血管瘤或色素沉着。发病有早有晚,可能在婴幼儿时已发病,有的在成年后才出现症状,这与脊柱裂引起一系列继发性病理变化以及脊髓栓系逐渐加重、发生缺血变化是一致的。按其临床症状,则有轻、中、重症之分,但有相当多的脊柱裂病人终生不发生症状。
1.轻症起病时的症状有下肢力量弱,轻度肌萎缩、麻木、遗尿,有时表现为腰痛或腿痛。多为一侧下肢受累,但也有两下肢同时发生肌无力者。检查发现呈周围性神经损害的表现,即肢体肌张力低,弛缓性轻度肌肉无力,下肢及会阴部浅、深感觉减退。
2.中症上述运动与感觉障碍较为明显,常见有马蹄内翻足畸形,有时出现腰痛、坐骨神经痛或伴发尿失禁。
3.重症下肢表现明显肌力减退,甚至瘫痪;感觉亦明显减退或消失,常并发神经营养性改变、下肢远端发凉、发绀,出现营养性溃疡。有的在骶尾部也常发生营养性溃疡,骶神经分布区皮肤感觉障碍明显。久之下肢呈现失用性萎缩,跟腱反射消失或发生挛缩。足畸形可出现仰趾足、弓形足、足内翻或外翻。部分患者表现为完全性截瘫及尿失禁,也有的为大便、小便均失禁。少数伴有椎间盘突出或腰椎滑脱,尚见有因脊髓栓系引起上肢症状者。
根据局部皮肤多毛、紫斑、小凹、色素沉着等临床表现,结合神经损害之症状,诊断多无困难。尤其是长期遗尿或发生明显尿失禁者,应多考虑为脊柱裂所引起。脊椎X线平片和CT与MRI扫描有助于疾病的诊断。
病因
(一)发病原因
世界各国自20世纪60年代起就陆续开展对脊柱裂和无脑畸形为主的神经管畸形的病因研究,20世纪70年代,通过一系列动物实验、临床观察和流行病学综合性研究,各国学者公认,神经管畸形是遗传因素和环境因素共同作用的结果,其中环境因素包括妊娠早期遭受如放射线、毒物、激素类药物、缺氧酸中毒等不良刺激。20世纪80年代的研究结果认为,妇女怀孕早期体内维生素缺乏可能是神经管畸形发生的主要原因,他们观察发现处于贫困和营养不良状态的妇女怀孕后,神经管畸形的发生率较富裕和营养状态良好的妇女要高的多。20世纪90年代最新的研究结果表明,妇女怀孕早期体内叶酸缺乏是神经管畸形发生的主要原因。遗传因素也是神经管畸形发生的原因,有人统计有8%~20%的病儿父母罹患隐性脊柱裂。曾有人用酸化培育受精卵蝌蚪液体,结果获得青蛙的脊柱裂动物模型。但目前对其发生机制了解得还不清楚。有神经管畸形家族史的家庭或夫妇,具有不明原因流产、早产、死产、死胎的家庭或夫妇、有致畸或放射性物质接触史的夫妇、近亲结婚的夫妇、已确定或可能为遗传病致病基因的携带者等,他们的后代发生神经管畸形的危险性较大。
(二)发病机制
神经管的闭锁,从胚胎17~19天开始大约需要10天左右的时间,由中胚叶分化成的椎板与肌板,共计42对体节中,从第4~7体节逐渐愈合,首先在24天(23~26天)左右前神经孔闭锁,随后26天(26~30天)左右,后神经孔闭锁。在这期间神经管与皮肤外胚叶间存在的中胚叶组织(体节)形成脊椎骨,全椎弓愈合完成是在胚胎8周左右。随后神经胚形成结束,覆盖皮肤外胚叶的前神经管进一步向尾端延长。在这期间以神经外胚叶和中胚叶为起源,位于前神经管尾端的未分化的细胞集团中产生空泡化。随后这些空泡逐渐融合,形成管状结构(后神经管的形成)。两神经管最终结合,进一步形成一条脊髓原基,这个过程称为造管。脊椎管内充满的脊髓原基在胚胎40~48天,脊髓尾端发生退行变性,形成脊髓终丝。脊髓的退行变性在生后依然进行,其结果是位于终丝头侧部的脊髓圆锥的位置渐渐上移,使在胚胎56~60天左右位于骶2~3平面的脊髓圆锥,在胚胎20周时位于腰4,在出生时位于腰2~3,在成人时达到腰1~2的水平。
人的中枢神经系统,即脑和脊髓,在胚胎的第1个月开始发育;在胚胎第2周,背侧形成神经板。神经板两侧凸起,中间凹陷,两侧的凸起部分逐渐在顶部连接闭合,而在胚胎第3~4周时,形成神经管。如果神经管的闭合在此阶段被阻断,则造成覆盖中枢神经系统的骨质或皮肤的缺损。神经管的最前端大约在胚胎第24天闭合,然后经过反复分化和分裂最终形成脑。神经管尾端后神经孔的闭合发生于胚胎的第27天而分化发育成脊椎的腰骶部。神经管及其覆盖物在闭合过程中,出现的异常成为神经管闭合不全。神经管闭合不全最多发生在神经管的两端,但也可能发生神经管两端之间的任何部位。若发生在前端,则头颅裂开,脑组织被破坏,形成无脑畸形。若发生在尾端,则脊柱出现裂口,脊髓可以完全暴露在外,也可能膨出在一个囊内,称之为脊柱裂和脊膜膨出。
隐性脊柱裂的病理形式很多。单纯的隐性脊柱裂不合并其他的脊髓或神经病变,椎管内容不向外膨出。在脊背部的外表,多为正常的皮肤,有时局部有毛发增生,色素沉着斑,片状毛细血管瘤,皮肤小凹,皮肤瘘口等。上述这些复杂的病理改变,常造成脊髓栓系与受压。
检查
无特殊异常表现。
1.脊椎X线平片摄腰骶椎前后位和侧位像,显示椎弓根增宽,椎板缺损,棘突缺如,有时呈现多处脊柱裂,或同时合并椎体畸形,脊柱侧弯等。
2.CT与MRI扫描特别是MRI对脊柱裂合并脊髓栓系的诊断更为明确。多能显示脊髓末端位置下移,达到腰骶交界或骶管内,局部存在粘连等征象。如今脊髓造影已被MRI所代替。
鉴别
隐性脊柱裂需与腰椎间盘突出、腰肌劳损、肌痛、脊髓肿瘤等鉴别。成人发病者还需与椎管狭窄症等鉴别。采用CT、MRI扫描可以明确诊断。CTM检查更能够反映脊髓的形态变化及程度,但与造影剂影像重叠,有时难以反映致压物的部位、形态及大小,尤其对骨化程度及神经组织的观察仍欠满意。
并发症
复杂的隐性脊柱裂常同时存在脊髓或神经发育异常,如局部瘢痕、粘连、终丝肥厚与增粗,使脊髓固定在椎骨上而限制了脊髓在发育中的上移,或同时有软骨瘤、脂肪瘤、表皮样囊肿、皮样囊肿、畸胎瘤、蛛网膜囊肿、脊髓末端空洞症、神经根憩室形成、脊髓内胶质增生或中央管扩张等情况。有时合并半椎体、脊柱侧弯、椎间孔与肋骨发育畸形。
治疗
(一)治疗
对于脊柱裂引起的脊髓栓系综合征者,均适合于手术,而且提倡尽早地予以手术治疗。只有手术解除脊髓栓系的因素,才能取得治愈、好转的机会。以往不少人将脊柱裂合并尿失禁或大小便失禁以及兼有下肢瘫痪者,视为手术禁忌证,亦即认为此类严重病例为不治之症。然而近20余年来积累了大量病例的临床处理经验,主张采取积极的手术态度,即使已有大小便失禁或下肢瘫痪者,也应争取手术。因此,对一些重症病例也不宜轻易放弃治疗。
1.手术治疗儿童多用基础麻醉加局麻,个别采用气管插管全麻;成人用强化麻醉加局麻,或采用硬脊膜外麻醉。均在俯卧位下手术。无论病变在颈段、胸段或腰骶段,都使用棘上直切口,以利于向脊柱裂病变节段之上与下方扩大椎板切口。
以腰骶部隐性脊柱裂合并脊髓栓系为例,棘上直切口自腰4至骶椎中部,按通常之椎板切开手术方法,分开椎旁肌肉,显露出缺裂之椎板。再按具体病理情况进行处理。
2.手术操作步骤可先从正常部位的中线切开,然后进入畸形部位。手术操作一般按以下步骤进行,即:
(1)扩大椎板减压,向上多切除1~2个椎板,向下至显露出终丝。将局部存在的一切造成对硬脊膜囊、脊髓、马尾神经牵拉、压迫的异常骨性、软骨及软组织予以切除。切除增厚的黄韧带,将外终丝切断,但需判明终丝两侧之骶神经,切勿损伤这些神经。如果脊髓栓系为来自硬脊膜外、硬脊膜囊与脊髓的压迫因素原因已经解除,手术即可终止,无需再切开硬脊膜探查。
(2)如见瘢痕组织、脂肪瘤或椎管内脊膜膨出囊连至脊膜囊内,必须继续切开硬脊膜囊作深部探查,并将瘢痕切除,分离粘连,摘除囊肿,使脊髓及神经根获得松解。如有脂肪瘤包绕脊髓与神经,尤其是脂肪与神经组织交融难分、即使在显微镜下也难以分出正常脊髓境界时,则只能作脂肪瘤的次全切除,避免破坏脊髓的血液循环或直接伤及脊髓与神经。若发现存在先天性皮样囊肿或肿瘤,应尽可能地将其予以切除。
(3)缝合硬脊膜、椎板缺损区不需植骨和作椎板固定术。其外可敷盖吸收性明胶海绵(明胶海绵),仔细止血后,按层缝合伤口。可在硬脊膜外留置引流管,24~48h后拔除。通常中、重症病人经手术治疗后,约半数病人可达到显效。
手术后采取俯卧或侧卧位姿势一周,有明显尿失禁者,宜进行导尿术,保持手术部位的清洁、卫生;对幼儿应严防大小便的污染,酌用抗生素预防感染。伤口拆线后可增加康复治疗项目,如理疗、针灸、按摩及肢体功能锻炼等,并应用神经营养药物,以促进神经功能的早日恢复。
(二)预后
手术解除脊髓栓系的因素,可以得到治愈、好转。合并尿失禁或大小便失禁以及兼有下肢瘫痪者,其中一部分病例手术后可得到康复或有所好转。现代显微手术方法的广泛应用,更增加了手术的疗效。
预防
预防:平素应嘱病人起居有节,保持心情舒畅,正确对待疾病,树立战胜疾病的信心由于隐性脊柱裂是遗传因素和环境因素共同作用的结果,需注意防止环境因素如妊娠早期遭受如放射线、毒物、激素类药物、缺氧酸中毒等不良刺激。妇女在怀孕早期补充体内维生素均有助于隐性脊柱裂的预防。
相关疾病
中毒 脊柱裂 颅裂 脊膜膨出 血管瘤 尿失禁 脂肪瘤 早产 死胎 流产 营养不良 脊柱侧弯 腰椎间盘突出症 腰痛 腰肌劳损 坐骨神经痛 截瘫 性病 麻醉 脊髓栓系综合征 瘫痪 脊髓肿瘤 皮样囊肿
