介绍
色素失调症(incontinentiapigmenti)又称Bloch-sulzberger病或Blcoh-si-ments病综合征。为一种遗传性疾病。在发生红斑、水疱、疣状或炎症改变后,出现色素性皮损,可伴有眼、骨和中枢神经系统缺陷。
最新文章
症状
出生后不久即发生,罕见2个月后发病。损害分3期:红斑及大疱;丘疹和疣状损害;色素沉着。它们的发生不规则,可相互重叠,分布多样化。
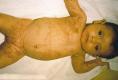
皮损的突出表现为灰褐色和黑灰色斑点,呈线状、水滴状、螺纹状和大理石纹状等不规则形,排列无次序,常分布于躯干、上臂、大腿部位(图1)。在色素斑出现前,常有大疱,尼氏征阴性,此起彼伏,水疱消退后可见红色光滑的结节和斑块。皮损中可出现蓝到棕色的泼溅样色素沉着斑。色沉斑持续数年后可渐消退,直至20~30岁才觉察到,有的伴有萎缩和硬化。
患儿常伴有假性斑秃、甲营养发育不良、甲缺损、甲畸形,眼部损害约占30%,表现白内障、斜视、视神经萎缩,蓝色巩膜、渗出性脉络膜炎等;骨骼异常少见,如并指(趾)、多肋、骨瘤、偏身萎缩及牙齿缺陷;出牙延迟或部分无牙。中枢神经系统也可受累,表现智力发育迟缓、小头畸形、癫痫、痉挛性四肢麻痹,短臂、短腿等。
根据临床表现,皮损特点,组织病理特征性即可诊断。
病因
(一)发病原因
是一种性联X连锁的显性遗传病,女性多于男性。
(二)发病机制
性联X连锁显性遗传的女性的异常基因,只位于2个X染色体之一,致病基因可被另一个正常X基因保护,因而病变不严重,存活率高。男性只有1个X基因,如是致病基因,病变较重,常死于宫内。常有近亲结婚及家族遗传史。
检查
组织病理:小疱位于表皮内,伴海绵形成,有多数嗜酸性粒细胞,水疱间表皮常含有小团的表皮细胞,呈涡轮状排列及散在的大的嗜酸性透明胞浆的角化不良细胞,真皮浸润如表皮,疣状增生处表现棘层肥厚不规则的乳头瘤样增生及角化过度,色素沉着处真皮上层噬黑素细胞增多,内含大量黑素颗粒,基底层黑素颗粒减少,有空泡变性。
鉴别
1.Franceschetti-Jadassohn综合征有人认为该病为色素失禁症的异型,色沉呈网状,无水滴状,早期无水疱,疣状损害。亦无齿和眼部损害。
2.脱色性色素失禁症多见女孩,无色沉斑,只有减色斑,前期无水疱、无炎症,无家族遗传史。
3.色素性荨麻疹又称肥大细胞增多症,该病色素斑不呈水滴状,刺激色素斑可出现风团,皮肤划痕征阳性,组织病理显示,真皮内可见大量肥大细胞。
并发症
本病属于染色体遗传性疾病,通常在近亲结婚,以及父辈患有染色体疾病遗传所致,且本病可形成红斑、水泡、感染等并发症,皮肤完整性被破坏,故可因患者抓挠诱发皮肤细菌感染或者真菌感染,通常继发于体质低下,或长期使用免疫抑制剂以及有灰指甲等真菌感染的患者,如并发细菌感染可有发热、皮肤肿胀、破溃及脓性分泌物流出等表现。严重病例可导致脓毒血症,故应引起临床医生的注意。
治疗
(一)治疗
本病的色素斑通常可自退,一般无需特殊治疗。对早期水疱炎症期可应用皮质类固醇激素。对于皮肤黏膜有感染的患者,可给予百多邦软膏涂擦治疗,全身感染患者,可予口服抗生素或者静脉使用抗生素进行治疗。
(二)预后
色沉斑持续数年后可渐消退,直至20~30岁才觉察到,有的伴有萎缩和硬化,本病比较迁延。
预防
本病属于染色体疾病,导致染色体畸形改变的病因不明确,可能和环境因素、遗传因素、饮食因素以及孕期的情绪、营养等具有一定的相关性,故本病无法直接预防。孕期应做到定期检查,若孩子有发育异常倾向,应及时做染色体筛查,明确后应及时行人工流产,以避免疾病患儿出生。
